INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease associated with cognitive decline, and is the most common form of dementia in the elderly.1 In addition to the serious cognitive impairment including memory impairment, AD is accompanied by several neuropsychiatric symptoms which are also equally as important as memory decline in the clinical features.2 Among the neuropsychiatric symptoms, sleep disturbances such as falling asleep, maintaining nocturnal sleep are known as the most frequent and serious behavioral symptom in AD associated with agitation, confusion, anxiety, aggressiveness falling down and cognitive declining.3 In these regards, understanding the neurobiological mechanisms would be very important to manage the sleep disturbances and their associated clinical consequences in AD subjects. To date, several previous studies reported an increase in the sleep modifications commonly observed in normal aging: loss of sleep continuity and a reduction in stages 3 and 4 and rapid eye movement (REM) sleep, shortening of REM sleep periods, trend towards a bimodal (afternoon and night) circadian partition of sleep.4 These may result in a positive-feedback loop, whereby poor sleep contributes to amyloid deposition, and amyloid plaque formation disrupts sleep through effects on sleep-promoting brain regions such as brain stem, hypothalamus and several prefrontal regions.5 Despite their clinical impact, there has been paucity of neuroimaging studies on the sleep disturbances of AD subjects. Only a functional neuroimaging study using single photon emission computed tomography (SPECT) showed increased perfusion of right middle prefrontal cortex in the AD subjects with sleep disturbances, compared with AD patients without sleep disturbances.6 As the magnetic resonance imaging (MRI) is known to well reflect disease progression along with AD,7 the identification of the structural abnormalities might be needed to understand the precise neurobiological mechanisms of sleep disturbances of AD.
The aim of this study is to investigate the neuronal substrate of insomnia associated with AD subjects. We hypothesized that the hypothalamus and prefrontal cortices might be involved in the sleep disturbances of AD.
METHODS
Subjects
Sixty subjects took part in this study [30 with AD and 30 healthy elderly controls (EC)]. All AD patients 1) fulfilled the National Institute of Neurological and Communication Disorders and Stroke/Alzheimer Disease and Related Disorders Association criteria for probable AD8 and 2) had a score on the Clinical Dementia Rating Scale ≥ 1.9 We excluded from the study subjects who had other neurological or psychiatric conditions (including other forms of dementia or depression) and those taking any psychotropic medications (e.g., cholinesterase inhibitors, antidepressants, benzodiazepines, and antipsychotics). The study was conducted in accordance with the ethical and safety guidelines set forth by the local Institutional Review Board of the Catholic University of Korea. Informed consent was obtained from all subjects and their guardians participating in the study. All subjects were right-handed. Cognitive functions were evaluated with the Korean version of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD-K), including Verbal Fluency, a 15-item Boston Naming Test (BNT), Mini Mental Status Examination (MMSE), Word List Memory (WLM), Word List Recall (WLR), Word List Recognition (WLRc), Constructional Praxis, and Constructional Recall (CR).10 The severity of sleep disturbance was assessed with the sleep disturbance domain of the Neuropsychiatric Inventory (NPI),11 measures the presence of sleep disturbances including difficulty falling asleep, nighttime awakenings, nighttime wandering, excessive sleep during the day, and early awakenings. Patients were characterized as having sleep disturbance if they scored 1 or greater on NPI sleep disturbance scales.
MRI Acquisition
All participants underwent MRI scans on a 3-Tesla whole body scanner equipped with an 8-channel phased-array head coil (Verio, Siemens, Erlangen, Germany). The scanning parameters of the T1-weighted three-dimensional magnetization-prepared rapid gradient-echo sequences were as follows: echo time = 2.5 ms; repetition time = 1900 ms; inversion time = 900 ms; flip angle = 9°; field of view = 250 × 250 mm; matrix = 256 × 256; and voxel size = 1.0 × 1.0 × 1.0 mm.
Image Processing
The image preprocessing and analysis steps were done in statistical parametric mapping (SPM) 8 (Wellcome Trust Centre for Neuroimaging, London, UK; http://www.fil.ion.ucl.ac.uk/spm) using the voxel based morphometry (VBM) 8 toolbox (http://dbm.neuro.uni-jena.de/vbm). We utilized the optimized VBM process12 which included 1) segmentation and extraction of the brain in native space, 2) normalization of the images to a Montreal Neurological Institute standard space, 3) segmentation and extraction of the normalized brain (extraction is repeated to ensure that no non-brain tissues remain), 4) modulation of the normalized images to correct for tissue volume differences due to the normalization procedure, and 5) sample homogeneity was checked to identify any outliers in the study population. The gray matter probability values were smoothed using an 8-mm full-width half-maximum isotropic Gaussian kernel. The smoothed gray matter images were analyzed with an analysis of covariance model using SPM8. Age, gender, and total intracranial volume were included as covariates.
Total intracranial volume can be approximated by calculating the sum of volumes of gray matter and white matter.
Statistical Analysis
Statistical analyses for demographic data were performed with the Statistical Package for the Social Sciences (SPSS) software (version 12.0; SPSS Inc., Chicago, IL, USA). Assumptions for normality were tested for all continuous variables. Normality was tested using the Kolmogorov-Smirnov test. Two-sample independent t-test was used to assess potential differences between the control group and the AD group for all continuous demographic variables, and chi-square tests for categorical variables. All statistical analyses used a two-tailed α level of 0.05 for defining statistical significance.
The general linear model (GLM) was used for measuring the group differences of the gray matter volume. In addition, the GLM was also used for measuring the correlation between the gray matter volume and the NPI sleep disturbance sub-score in the AD group. We controlled the effect of age, education, and gender from the all GLM analysis used. The threshold was set at p < 0.05 [false discovery rate (FDR)] to control for multiple comparisons.13
RESULTS
Demographic Data
Table 1 shows the baseline demographic data in our different subject groups. No significant differences in sex, age, and education were observed between the AD group and the EC group. Compared with EC, patients with AD showed significantly poorer performances in BNT, MMSE, WLM, WLR, WLRc, and CR on CERAD-K neuropsychological tests (p < 0.05). All of the subjects had sleep disturbances which was characterized as difficulty falling asleep (47%), nighttime awakenings (30%), nighttime wandering (20%), excessive sleep during the day (7%), and early awakenings (10%).
Group Analysis
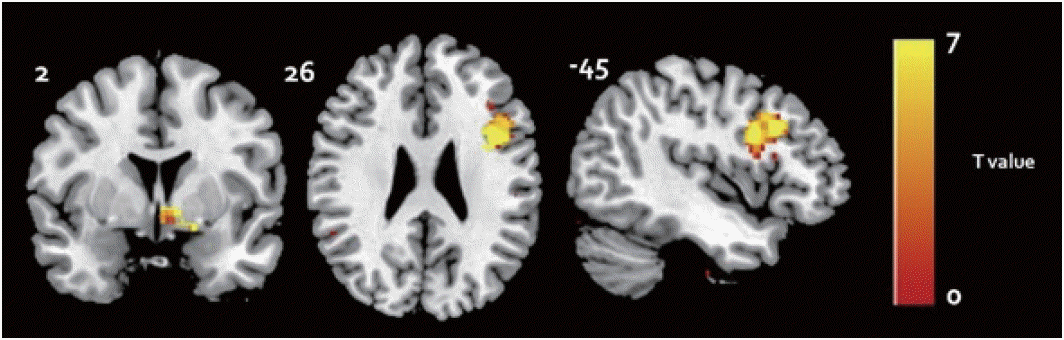
A group comparison analysis of the regional gray matter volumes between the AD and the EC group showed a significant reduction in the gray matter volumes of the AD group in the left medial temporal, the posterior cingulate, the precuneus, the dorsolateral prefrontal cortex (DLPFC), and anterior cingulate as compared with the EC group (p < 0.05 FDR corrected) (Table 2, Fig. 1).
DISCUSSION
To the best of our knowledge, it is the first study to elaborate the relationships between gray matter volumes and sleep disturbances in AD.
We found the hypothalamus and left DLPFC were negatively correlated with the gray matter volumes in the AD group, which was in line with the previous functional study using SPECT.6 They reported that AD subject with sleep disturbance showed increased brain perfusion in right DLPFC as compared with the AD subjects without sleep disturbances.6 They suggested this relative hyperperfusion of the DLPFC might be associated with a decreased ability to further deactivate the DLPFC resulting in sleep disturbance from abnormal slow wave sleep in AD subjects. During the normal sleep period, DLPFC is known to be deactivated from waking to non-REM sleep, as evidenced by positron emission tomography (PET) study.6,14 Several previous PET studies using H2[15O] showed, the prefrontal areas reactivated during REM sleep include anterior cingulate, caudal orbital and medial prefrontal cortices,15 but the DLPFC remained deactivated.14 In these regards, prefrontal deactivation pattern is increasing with deepening of non-REM sleep, and maintained in the transition from non-REM sleep to REM sleep. On the other hand, with the onset of REM sleep, portions of the ventromedial, limbic-related prefrontal cortex and closely associated medial subcortex and cortex are reactivated, sometimes to levels that exceed those of waking,16 but the DLPFC remains relatively deactivated with inhibition of acetylcholine.6,17
Our findings expand these previous neuroimaging to association of sleep disturbance of AD with gray matter structural abnormalities including DLPFC and hypothalamus. To the best of our knowledge, there has been no study on association between sleep disturbance and gray matter volume in AD. As compared with medial temporal structures including the entorhinal cortex, the DLPFC is known to be pathologically spared during the early stage of AD trajectory.18,19 However, as the DLPFC is responsible for the various attention require process and behaviors including executive function, planning, self awareness, initiation, and mood regulation.20 Therefore, the damage to the DLPFC by AD pathology might produce various cognitive and behavioral disturbances.
The hypothalamus is very well known as key contributor to the modulation of sleep wake cycle. The hypothalamus can be grossly separated into five groups of neurons–the hypocretin-expressing neurons of the lateral hypothalamus, the suprachiasmatic nucleus (SCN, location of the circadian pacemaker), histamine-expressing neurons, the ventrolateral preoptic nucleus, and temperature-sensitive neurons in the anterior hypothalamus. Several postmortem studies showed the degeneration of the hypothalamus-especially SCN-in AD patients. The loss of SCN arginine vasopressin (AVP) neuron and its associated accelerated rhythmicity was reported. In addition, loss of neurotensin-expressing neurons in the SCN of AD patients is also reported along with increased astrocytes.21 As AVP, and neurotensin are known to alter SCN neuronal function,22,23 their loss during AD might result in the circadian dysregulation of AD patients. The previous studies reported that neuropeptide alterations in the SCN occur at early stages of AD and might preface cognitive decline.24 In addition to the SCN, the cholinergic basal forebrain might be important for circadian rhythm disturbance in AD. The basal forebrain is connected to the SCN and modulates circadian rhythms by cholinergic neuronal activity.25,26 The degeneration of basal forebrain and subsequent loss of cholinergic cells during the AD process might result in the circadian rhythm disturbances. In the previous animal study, lesion of the cholinergic projection to the SCN in rats leads to alterations in the phase-shifting effects of light on circadian rhythms.25 As noted above, the DLPFC is directly deactivated by acetylcholine activity and acetylcholinesterase activity of the pedunculopontine and laterodorsal tegmental nuclei may be abnormal in AD patients with sleep disturbance compared to those without during the REM sleep.6 Taken these together, our results imply that the degeneration of the hypothalamus and the DLPFC in AD might be the core neuronal substrate of the sleep disturbance in AD. Furthermore, as we recruited the drug naïve subjects with AD, we could show the more precise neuronal substrate of sleep disturbances of AD without influence of acetylcholinesterase inhibitors.
The limitations of our study were as follows. First, as nearly all of the AD subjects had a certain degree of sleep disturbances; we could not show the group differences between the AD subjects with and without sleep disturbances. Further study with the AD subject without sleep disturbance might be needed for the precise neurobiological mechanisms of sleep disturbances of AD. Second, although we used the sleep disturbance domain of the NPI, this sub-scale could not precisely characterize the sleep disturbances in detail. Hence, the results of our study could be over speculative. In the future, the more precise characterization of sleep disturbances with polysomnography might be needed.
In conclusion, we showed the aberrant gray matter volumes of the DLPFC and the hypothalamus might be related to the sleep disturbances of AD patients. This distinctive association might be helpful to understand the detrimental clinical feature of sleep fragmentation of AD and develop the treatment strategy of serious behavioral disturbances originated from sleep disturbances in AD such as agitation and falling down.